
Whether you are a medical device company looking to implement a quality management system or a start-up that is just getting into the industry, you need a QMS that is compliant with ISO 13845.
In this article, we will go over our best tips and tricks for setting up or adapting your QMS to ISO 13485 standards.
1. Plan and define your project programme
When to start? How to start? Where to start? Read on to find out more as the answers to these commonly asked questions can differ from business to business.
1.1 When?
Ask anyone who has been a part of an ISO13485 QMS implementation project and we would all say, “the sooner the better”; there are good reasons for that. As soon as you are certain that your product is a medical device and you have classified it, you should start building or adapting your QMS to ISO 13485. Read our blog to find out more about how to classify medical devices.
It is important to understand that, in the medical device world, if an action such as a meeting or a verification test of your product is not documented, (i.e. not properly signed and saved as a record) it never happened!
To understand why you should start early and the consequences of not doing so, let us imagine that you do the opposite, which would be to fully design and develop your product first. You will then have a finished product, which you cannot take to market until you have circled back and retrofitted design and development records according to your QMS. This is time-consuming inefficient and expensive compared to following a compliant design and development procedure during product creation.
Furthermore, by following a compliant design control and product risk management procedure, you will design a safer medical device. You are also likely to avoid some extra time-consuming detours by planning in advance and ensuring no crucial parts of your product are left behind, such as labelling and packaging.
1.2 How?
It is important to let the people responsible for the process, and the ones who are actually working according to the process, be in charge of documenting how they work or how they plan to work. Then a QA (quality assurance) person or someone with deeper knowledge and training in ISO 13485 also needs to be involved to ensure regulatory compliance. For example, a perfect sub-team to create the supplier control procedure would be the manager responsible for the whole process, a specialist who will perform the day-to-day work and a QA specialist.
If you find it difficult to start from scratch then it can be a good idea to use a template created by a medical device QMS specialist, like MedQdoc. A template can be a good inspiration and help to ensure compliance. But, remember that a template is only a suggestion, a guide, and in the end, you need to describe your unique procedure and ensure it follows the applicable regulations. Find out more about MedQdoc’s full list of QMS templates here.
Depending on the size of your company, we would suggest appointing an allocated project leader that works very closely with the company’s management representative or, if you are very small, appoint the management representative as the project leader and drive the implementation as a project with clear goals and timelines.
1.3 Where?
If you are starting from a current QMS, possibly an ISO 9001 system or an older ISO 13485-compliant system that has not been maintained properly, I would start by doing a gap analysis of your current system towards ISO13485:2016. That way, you’ll get a good foundation for your project plan that focuses on filling out the current gaps.
If you are starting from scratch, the key process required to allow you to proceed is a document control procedure, including record management. From then on, everything you create will either be a controlling document / (a process/procedure description), or a record (documented proof that an activity within a process has been / carried out).
With that complete, where to start depends on how far you have come with your product and also what type of business you are.
Assuming that you are reading this when design and development are only in the concept phase, then our recommendation would be to start building design control, product risk management and supplier control procedures, and move on to production control and monitoring and measuring procedures later.
If you have already finished your design and are in the middle of selecting suppliers, then I would start with supplier control SOPs. Make sure to follow those as soon as possible and then maybe design control can wait for a little, since you are already too late and will have to do a catch-up recording.
2. Ensure commitment from your company and its top management

It is a common misconception amongst medical device customers that the QMS is something that the Quality director handles, a bunch of documents to be shown for auditors, and that everyday operation is something performed on the side.
If the same sentiment is shared within your company then the first step is to start by educating your colleagues on what it really means to implement a QMS according to ISO 13485, and the benefits it can bring to your product quality and business as a whole.
Start with the management team to get them completely on board first and then continue throughout your company.
Make sure that company management and all staff involved are committed, engaged, and truly believe in the benefits of a QMS.
3. Allocate time to the project

Take the time you need to set up the processes that you will follow. Preferably, describe what you are already doing and make sure to involve your ISO 13485 trained resources to ensure all necessary steps are included.
In this context – faster isn’t always better. It will be far more beneficial in the long run to involve the people who will be responsible for and work with the procedures on a day-to-day basis to ensure that they fully appreciate and understand the compliance activities of your business. By involving the staff who will be working with the QMS, you are implementing your QMS while creating it. So, while it may become a longer project if these individuals have little time to spare, the final product will be a functioning QMS.
Don’t believe anyone claiming that they can help you go from nothing to a compliant ISO 13485 QMS in 30 days. You will most probably end up with a paper product that is not an established QMS.
4. Allocate Resources – knowledge and competence

Apart from time, you need to have enough human resources allocated for an ISO 13485 QMS project to ensure that the correct knowledge and competence are present.
It is recommended that all management and most staff set time aside for an ISO 13485 QMS project and ring-fence some resources dedicated to this project for a period of time. It is also a good idea to ensure that it is not only people representing the quality department (the management representatives, QA specialists and/or quality consultants) that have allocated time.
Ensuring competence will look different from company to company. Maybe you already have experienced ISO 13485 experts within your organisation? Maybe you have allocated resources but they need a boost/training in ISO 13485? Maybe you need to use expert consultants to help drive training efforts within your company? Expert consultants, like MedQtech, specialise in offering medical device consultancy services that can help companies with training requirements and implementing their QMS.
5. Choose your QMS approach: electronic or paper-based
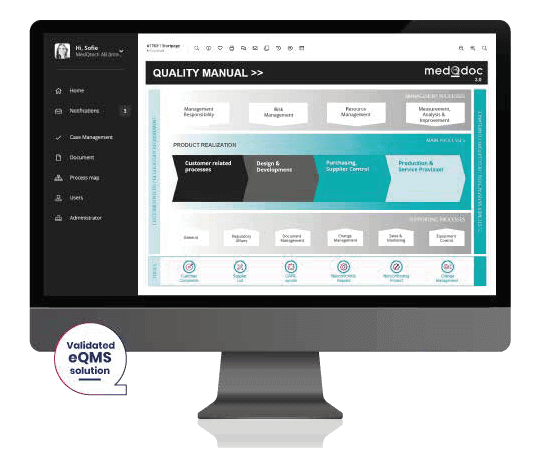
Implementing an electronic QMS brings many benefits, some of which are listed below. As experienced consultants in the medical device industry, we believe that the cost of an eQMS is quickly paid back in the form of company efficiency from handling fewer audit remarks and fewer resources engaged with simple but time-consuming administrative tasks.
The increase in remote working combined with the strict document control regulations in the medical device world can mean that obtaining signatures from multiple persons/functions within the organisation for every document could take days, or even weeks with a paper-based system. With an eQMS, it can be done within a minute regardless of where individuals are based.
Paper-based documents not only require handwritten signatures, but they also need to be stored in a fire-proof physical archive. They also require strict version control so that only the current version of the document is distributed within the organisation. All of this is managed automatically in an eQMS and can be done while working from a home office or while travelling.
Some may feel that upgrading their paper-based QMS to an eQMS is a big undertaking since it involves time-consuming administrative routes, which create obstacles to continuous improvements of your QMS. However, a paper-based QMS risks becoming just a pile of papers that is never used to create efficiency in your business and improve the quality of your medical device.
It is most often a good decision to implement an eQMS at the same time as your ISO 13485 compliant QMS. This way you don’t have to move your paper-based QMS at a later stage – you can do it as one project instead of two.
If you decide to implement an eQMS, we would strongly recommend using a system adapted for medical devices. With an eQMS solution, you can either buy a set of templates for the processes and records you need or alternatively, a few solutions provide them as an included feature. Such templates and structures are often a valuable help when setting up your processes and can really help shorten your ISO13485 compliance project. An example of an eQMS solution adapted to the medical device industry and regulations with integrated templates is MedQdoc.
6. Consider other applicable regulations and standards
If you are in the medical device industry, ISO 13485 is not the only regulation and standard to consider. Both ISO 14971 – regarding risk management to medical devices -and EN 62366 – covering usability engineering to medical devices – will almost always apply and should be integrated into your QMS.
If you are planning to market your product in Europe, you must at least consider Article 10 Chapter 9 in MDR regarding QMS. But it is also beneficial to integrate some of the other topics within your QMS, for example, post-market surveillance and clinical evaluation. If you are planning to market your product in the USA, you must follow 21 CFR 820 (QSR).
Whether you are an American or European company, or from elsewhere, our recommendation is to implement a QMS that is compliant with both QSR and ISO 13485. The differences are so small that accommodating both standards will take hardly any extra effort, and chances are you might expand your markets sooner than you think.
7. Carry out internal audits and management reviews
Regardless of what your QMS status is, two key processes that you really want functioning well are your management responsibility (including management review procedure) and internal audit processes. If these two processes are used and followed as intended by ISO 13485 and QSR, they will drive and improve your QMS, increase business efficiency as well as the quality of your medical device in the long run.
7.1 Internal Audits
In addition to checking for compliance, internal audit processes are effective tools for continuous improvement.
Don’t create a climate where the internal audit process is about finding and hiding faults. As it is easy to become blind to your own flaws, use the process as a guide to identify improvement opportunities and errors that you might have overlooked.
There are two key benefits of performing internal audits early according to your new process: firstly, it allows you to practice and find improvement opportunities within the internal audit process itself, and secondly, it allows you to improve and push all other processes within the QMS forward.
7.2 Management Reviews
Performing your first management review according to your new process at an early stage is a good idea – even if a lot of the topics get covered quickly. Getting in the habit of regularly reviewing your QMS, and the product and process data will benefit you later.
If you are new to performing ISO 13485 compliant management reviews, I would suggest you do them at least twice a year to get familiar and comfortable with the process.

8. Use your processes

We recommend that medical device companies use their processes as soon as possible rather than waiting until they are perfect before implementing them. It is better to release the first attempt and try to follow that. This is the best way to figure out what processes are and aren’t working for your company.
Continuous improvements and the risk management of processes are part of a well-functioning QMS. They are also great ways to set up well-functioning processes that increase the quality of the end-product as well as achieve regulatory compliance. A QMS with several processes with a high version/revision number is often a sign of a healthy, well-maintained system.
So, start following your processes at a very early stage and don’t be afraid to change them. Update them continuously when you figure out better and more streamlined ways of working.
Implementing and building a QSR/ISO 13845 compliant QMS needs a well-thought-out strategy and project plan – we hope that this guide has given you some helpful pointers on how to move forwards.
Some key takeaway points from this article are: get management and colleagues on board with the benefits of implementing an ISO 13485 project, allocate resources and time, document and use your processes as you build your QMS and regularly review and improve your system through management reviews and internal audit processes.
If you need any assistance with implementing a compliant QMS, please do not hesitate to contact our specialist MedQtech team.



MedQtech supports the medical
device industry to deliver effectively
